Toxicology Module
Greening Across the Chemistry Curriculum English | Versión en Español  | Versão em Português (Brasil)
| Versão em Português (Brasil) 
Biochemical Toxicology of Insecticides: The Road Towards Reduced-Risk Insecticides
Timothy D. Foley, Chemistry Department, University of Scranton
timothy.foley@scranton.edu
INTRODUCTION
The use of chemicals to control levels of crop-damaging insects has helped to increase agricultural efficiency during the past century. This practice has also been plagued by controversy. Insecticides have traditionally targeted nervous system functions that are common to many species including humans. The use of these neurotoxic insecticides has led to the indiscriminate killing of beneficial insects and posed a serious risk to other animals and humans from environmental exposures. Moreover, increasing evidence has suggested a possible link between human environmental exposures to insecticides and neurological diseases such as Parkinson's disease (1). Thus, there is a tremendous need to develop insecticides that display greater target selectivity and, thus, a reduced risk to non-target species and humans. In recent years, a novel class of bisacylhydrazine insecticides developed by Rohm and Haas Company (Philadelphia, PA) have been found to display highly selective toxicity by interfering with the unique physiology of insect growth. Two of these compounds have recently been designated as "reduced-risk" by the Environmental Protection Agency (EPA). On another front, an understanding of the more subtle biochemical mechanisms by which low levels of environmental chemicals (xenobiotics) may increase the risk for developing specific human diseases is rapidly emerging. This understanding should enhance our ability to screen for potential adverse health effects that are not readily predicted by traditional toxicological testing. The following toxicology module provides an overview of 1) nervous system function, 2) the primary mechanisms of traditional and bisacylhydrazine insecticidal actionand acute non-target toxicity, and 3) subcellular mechanisms of xenobiotic action that are distinct from these mechanisms but that are becoming recognized as relevant to human disease.
NERVOUS SYSTEM FUNCTION
SYNAPTIC TRANSMISSION
Nervous system function is mediated largely by the communication between the excitable (i.e., responsive to changes in ion levels) nerve cell and other excitable cells of an organism which may be either other neurons or muscle cells (2). Communication is initiated by the release of chemical messengers called neurotransmitters from a presynaptic nerve cell. The neurotransmitter diffuses across synapse (space) between the nerve cell and a contacted (postsynaptic) cell and binds to a receptor protein on the membrane of that cell (Fig. 1). Binding stimulates the opening of an ion channel (pore) in the membrane which facilitates the flux of specific ions (e.g., Na+, K+, Ca2+, or Cl- depending on the channel) across the membrane down their concentration gradients either into or out of the cell. Neurotransmitters may be excitatory orinhibitory depending on how they change the ionic charge on the inside of the cell which, in the absence of neurotransmitter, is negative with respect to the outside of the cell. Excitatory neurotransmitters depolarize the cell by promoting an influx of Na+ which, in some case, may be accompanied by Ca2+. This initial depolarization is propagated by the opening of voltage-sensitive Na+ channels along the axon (the skinny part) of the nerve cell and ultimately results in the opening of voltage-sensitive Ca2+ channels in the nerve ending. The influx of Ca2+ stimulates the release of neurotransmitter into the next synapse. In muscle cells, the influx of Ca2+ stimulates muscle contraction. In contrast to excitatory neurotransmitters, inhibitory neurotransmitters hyperpolarize the cell by promoting either an efflux of K+ ions or an influx of Cl- ions. This makes the cell more resistant to depolarization, Ca2+ influx, and release of neurotransmitters (in neurons) or contraction (in muscle cells).
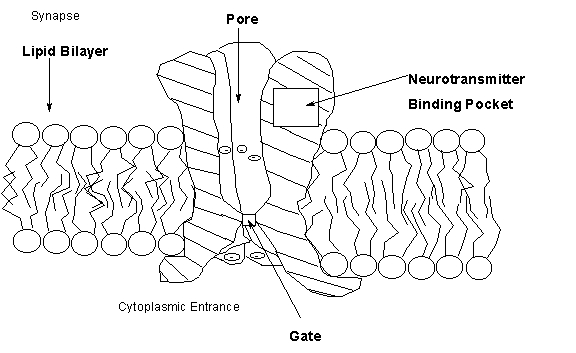
Figure 1. Neurotransmitter Receptor and Ion Channel
MECHANISMS OF ACUTE INSECTICIDE TOXICITY
ORGANOCHLORINES: EXCITOTOXIC INSECTICIDES
The organochlorine category of insecticides includes DTT (dichlorodiphenyltrichloroethane), lindane, and the cyclodienes (e.g., dieldrin, endrin, chlordane, and heptachlor) (3). These insecticides are generally very lipophilic and are not readily transformed to more hydrophilic metabolites and excreted. Thus, they are able to partition into fatty tissues and bioaccumulate. Indeed, DTT may remain in the body for years. Organochlorine insecticides generally act by promoting excessive increases in the excitability (i.e., sensitivity to depolarization) of neurons. This causes rapid and repetitive firing of neurons which manifests as tremors, hyperexcitability, convulsions, and eventual paralysis. This mode of neurotoxicity is called excitotoxicity. The mechanisms by which DDT, lindane, and the cyclodiene insecticides produce excitotoxicity are outlined below.
DTT
The lipophilic nature of DTT (Fig. 2) allows it to concentrate in the cell membrane and affect the function of membrane proteins (e.g., channels, receptors, transporters). The major mechanism by which DTT acts is thought to be by prolonging the opening of membrane-bound Na+ channels although it may also modify the function of several other membrane proteins (3). The effect of DTT on Na+ channel kinetics is temperature-sensitive and is not observed at temperatures above about 30 degrees Celsius. This temperature-dependence is believed to explain why DTT is much more toxic to colder-blooded insects, fish, and aquatic invertebrates than it is to humans (3). However, the environmental persistence, bioaccumulation, and non-target toxicity of DTT, brought to the Nation's attention by Rachel Carson's novel Silent Spring, led to DTT being banned for use in the U.S. in 1973 although human exposures may still result from products imported into the U.S.
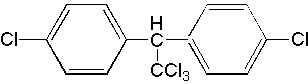
Figure 2. DDT
LINDANE and the CYCLODIENES
Lindane is the gamma isomer of hexachlorocyclohexane (BHC) (3). Cyclodienes are a class of organochlorine insecticides that are prepared from hexachlorocyclopentadiene by the Diels-Alder and subsequent reactions (3). They include dieldrin, endrin, chlordane, and heptachlor. Like DTT, lindane and the cyclodienes are very lipophilic. They resemble picrotoxin, an antagonist (inhibitor) of a postsynaptic receptor for the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). The binding of GABA to this receptor, called the GABA-A receptor, stimulates influx of Cl- ions which hyperpolarizes the cell and makes it more resistant to depolarization. Thus, these insecticides promote excitotoxicity by blocking the stimulation of Cl- influx by GABA. Lindane and the cyclodienes exhibit significant non-target toxicity. They are very toxic to non-target insects, fish, and birds. The cyclodienes are also toxic to other mammals.
ORGANOPHOSPHOROUS AND CARBAMATE INSECTICIDES: ACETYLCHOLINESTERASE INHIBITORS
ACETYLCHOLINE and ACETYLCHOLINESTERASE
Acetylcholine is a neurotransmitter that, upon release from neurons, stimulates the opening of a Na+ and K+ channel that regulates the function of the brain as well as the heart, lungs, and skeletal muscles (2). Acetylcholine signaling in the synapse is terminated by an enzyme acetylcholinesterase (AChE) which catalyzes the hydrolysis of acetylcholine to form inactive acetate and choline (Fig. 3) (2-4). AChE is a member of the serine esterase class of enzymes that contain an active site serine (Ser) in addition to histidine (His) and glutamate (Glu) amino acid residues that cooperate to catalyze the hydrolysis of acetylcholine. Briefly, H-bonding occurring between the Glu carboxylate group and the N-1 of the His imidazole ring enhances the ability of the N-3 of His to act as a base and abstract the H from the Ser hydroxyl group. This cooperation makes the Ser oxygen a stronger nucleophile which readily attacks the carbonyl carbon of acetylcholine. This reaction results in the formation of a tetrahedral intermediate that is likely stabilized by H-bonding in an "oxyanion hole" (Fig. 4). The more stable binding of the tetrahedral intermediate than acetylcholine itself to the enzyme active site is a major reason why AChE is able to catalyze this reaction. Collapse of the tetrahedral intermediate and release of choline leaves behind the acyl enzyme. The acyl enzyme is attacked by a water molecule causing the release of acetate (i.e., by hydrolysis) and the regeneration of the active site Ser which is now poised for another catalytic cycle.
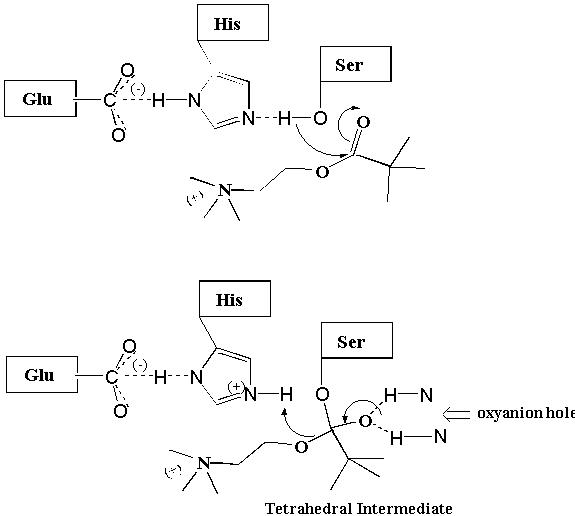
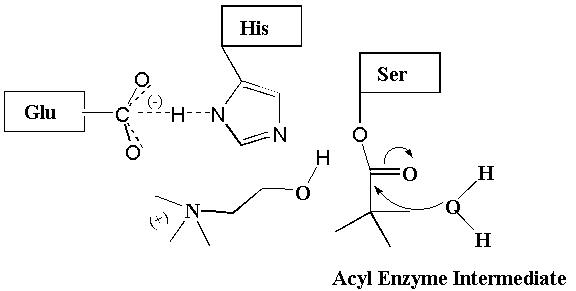
Figure 3. Acetylcholinesterase Mechanism
Organophosphorous (e.g., parathion and malathion) (Fig. 4) and N-methylcarbamate (e.g, carbaryl, aldicarb) insecticides are believed to act by inhibiting AChE activity (3,4). They do so by acting as pseudosubstrates and forming a covalent adduct with the active site Ser. This results in accumulation of acetylcholine in the synapse, overstimulation of AChE receptors and, ultimately, death by respiratory failure. Parathion and malathion become much more potent AChE inhibitors following oxidation in a reaction catalyzed by cytochrome P450 monooxygenases. Indeed, the sensitivity of an organism to organophosphorous compounds is greatly determined by the relative rates of oxidative transformation vs a hydrolytic conversion to less toxic species. Differential metabolism is believed to underlie the lower sensitivity of mammals to malathion. Oxidative metabolism replaces sulfur with the more electronegative oxygen. This increases the positive charge on the phosphorous atom and makes it more reactive towards the AChE Ser. Moreover, the rate of hydrolysis of the resulting phosphorylated enzyme is generally so slow that AChE is likely to be degraded and replaced by a newly synthesized enzyme before release of phosphate occurs. The replacement of AChE may occur with a half-life of 10-30 days so that repeated exposures to subtoxic doses of organophosphates may produce a cumulative response. The N-methylcarbamates are generally more readily reversible and, thus, less toxic to non-target organisms since repeated exposures are less likely to produce an additive effect.
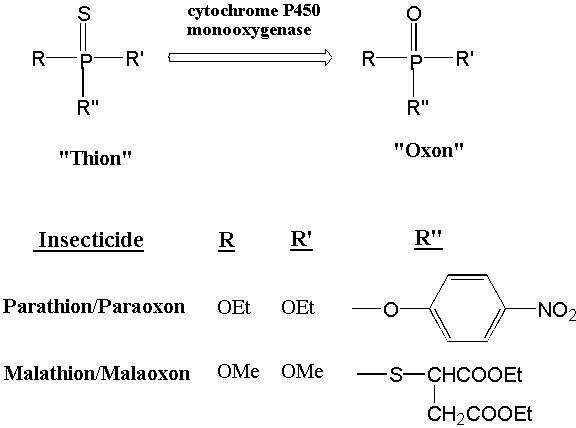
Figure 4. Organophosphorous Insecticides
BISACYLHYDRAZINE INSECTICIDES (GREEN CHEMISTRY)
In recent years, it has become apparent that more selective toxicity may be achieved by targeting the unique physiology of insect growth. The growth of insects is accompanied by the periodic shedding of their outer cuticle layers, a process called molting. Molting is initiated by an increase in the levels of the steroid hormone 20-hydroxyecdysone (20E) (Fig. 5) and terminates upon metabolism of 20E and a decline of 20E concentrations to basal levels (5). 20E acts by binding to an ecdysteroid receptor protein. The 20E-receptor complex directly activates the expression of genes that are involved in the molting process. In 1988, two papers in the journal Science reported that the bisacylhydrazine compound RH-5829 (Fig. 6), developed by scientists at Rohm and Haas Company, was a metabolically stable ecdysteroid receptor agonist (6,7). That is, it induced the molting process by apparently binding to the ecdysteroid receptor like 20E. However, because it is not rapidly metabolized, the levels of RH-5829 remain high and the target insects are unable to recover from a RH-5829-induced molt (5-7). They die of dehydration and/or starvation. Thus, RH-5829 produced a lethal molt by virtue of both its ecdysteroid agonist activity and metabolic stability. Since then, other more potent and selective bisacylhydrazine insecticides such as tebufenozide and halofenozide (Fig. 5) have been developed. Notably, tebufenozide and halofenozide have been designated "reduced risk" pesticides by the EPA and, for their development, Rohm and Haas received a Presidential Green Chemistry Challenge Award. Indeed, they exhibit a remarkable target selectivity with little or no toxicity reported for many other beneficial insects. Specifically, tebufenozide kills caterpillars while halofenozide kills beetle larvae, cutworms, and webworms.
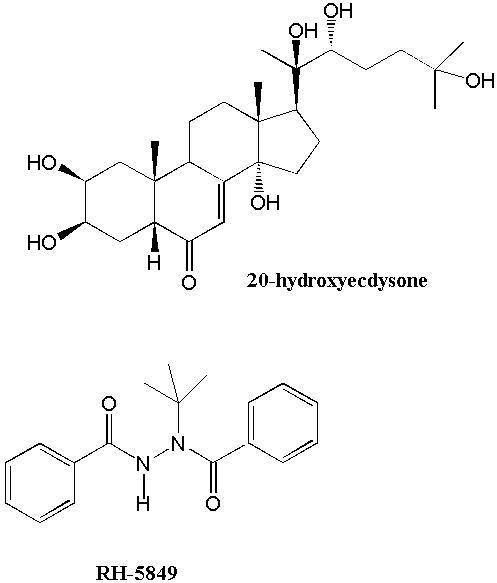
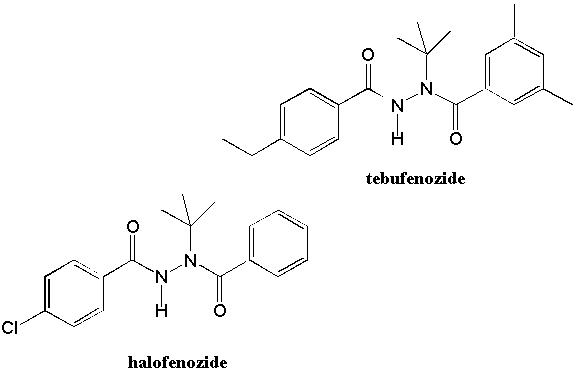
Figure 5. 20-Hydroxyecdysone and the Bisacylhydrazine Insecticides
The basis for the insecticide selectivity exhibited by the bisacylhydrazines is not entirely clear since all molting insects contain 20E and ecdysteroid receptors. Selectivity appears to be partly attributable to differences in the binding affinity of the bisacylhydrazines for the ecdysteroid receptors of different insects (5). It also may involve differences in bisacylhydrazine metabolism and transport among different species of insects (5). Of course, it is also plausible that the relatively small differences in binding affinities of the bisacylhydrazines for the receptors of different insects may reflect much larger differences in the receptor proteinconformation.
The acute toxicity of the bisacylhydrazines to higher organisms has been reported by Rohm and Haas to be very low (5). However, they are significantly lipophilic and may affect membrane structure and function. In fact, they have been reported to produce excitotoxicity in some insects by partitioning into membranes and blocking K+-channels (8), a mechanism that appears to be independent of ecdysteroid activity. Thus, the potential for ecdysteroid-independent toxicity and, indeed, neurotoxicity should be recognized and further explored. Nonetheless, the bisacylhydrazine insecticides highlight a novel strategy for developing insecticides with greater selectivity. Furthermore, they are being used as tools to further understand the unique biochemistry and physiology of insects which should identify other molecular targets for insect-selective toxins.
ADDITIONAL CONSIDERATIONS: ENVIRONMENTAL EXPOSURES AND HUMAN DISEASE
Targeting of the unique physiology of insects, as demonstrated by the bisacylhydrazine insecticides, represents a major advancement in the design of reduced-risk insecticides. However, it is important to recognize that the label of "reduced-risk" generally refers to a lower risk of gross symptoms of toxicity (e.g., death, convulsions, tumors, respiratory distress) in animal models (usually rodents) following acute (immediate) or short-term (< year) exposures. Thus, the information that can be obtained concerning the relationships between environmental exposures and human diseases (e.g., cancer, heart disease, and neurological diseases), which may develop over decades, is very limited. The mechanisms by which exposure to xenobiotics may increase the risk for developing human diseases are becoming more apparent and may be very distinct from the mechanisms of acute toxicity outlined above. Three such mechanisms are outlined below.
MITOCHONDRIAL DYSFUNCTION
Mitochondria are the sites of electron transport (Fig. 6) and the coupled synthesis of the major part of ATP produced in our cells (9). Mitochondrial dysfunction results in lower ATP production, increased generation of toxic forms of oxygen (e.g., superoxide, hydrogen peroxide), and, recently, has been implicated as an initiator of the orchestrated form of cell suicide known as apoptosis (10). Moreover, increasing evidence supports the notion that aging-related mitochondrial dysfunction may play an important role in the development of human neurological disease (10). Thus, findings that a wide range of chemicals, including insecticides, perturb mitochondrial function have profound implications for our understanding of potential toxicities and relationships of chemical exposures to human disease. Indeed, some organophosphorous compounds including parathion induce changes in mitochondrial membrane permeability and inhibit mitochondrial function (11). Furthermore, some chemicals that have been used as insecticides such as rotenone, dinitophenol, and cyanide inhibit mitochondrial function at well established sites and are used as tools to study mitochondrial function (9). In addition, members of the pyrethroid family of insecticides, which were not discussed above, have recently been found to potent inhibitors of mitochondrial complex I (12), the same target as the chemical MPP+ which induces Parkinson's-like symptoms in humans (10).
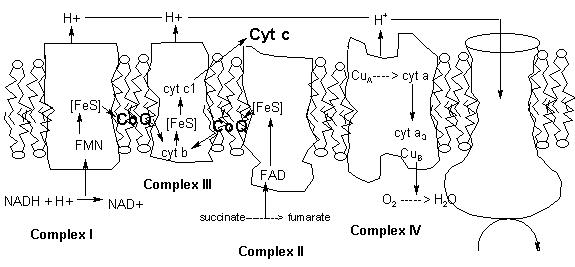
Figure 6. Mitochondrial Electron Transport
ALTERATIONS IN CYTOCHROME P450 MONOOXYGENASES
The cytochrome P450 monooxygenase (CYP) enzymes are the major enzymes involved in the initial phase (phase I) of xenobiotic metabolism (4), often referred to as biotransformation to distinguish it from intermediary (energy) metabolism. CYPs generally oxidize xenobiotics (Fig. 7), by hydroxylation for example, which often renders them substrates for conjugation reactions with large hydrophilic metabolites and, ultimately, promotes excretion in the urine. Importantly, however, biotransformation by CYPs can increase as well as decrease the toxicity of a foreign substance (4). Furthermore, although the physiological role of these enzymes is unclear, they may be important for removal of endogenous toxic compounds that are generated during normal cellular metabolism. Many pesticides including organochlorines and organophosphorous compounds have been shown to inhibit the activity and/or alter the expression of various CYP isoforms (13). For example, parathion inactivates the CYP3A4 isoform during its oxidative biotransformation (14). These changes may increase the sensitivity of cells to reactive endogenous metabolites or other xenobiotics (4). Thus, it has been postulated that inhibition of CYP activity by organophosphorous compounds could contribute to the development of Parkinson's disease by rendering neurons more sensitive to toxic metabolites of neurotransmitters that are normally metabolized by CYP (1)
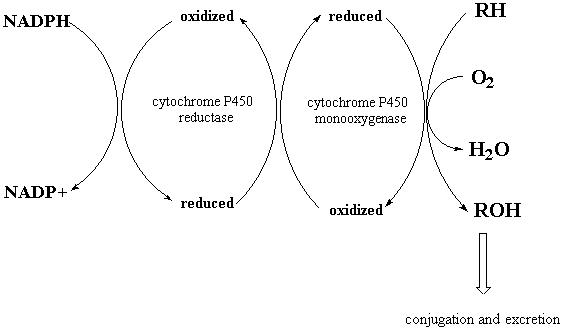
Figure 7: Hydroxylation of a Substrate by Cytochrome P450 Monooxygenase
DISRUPTION OF HORMONE BALANCE
Hormones play a central role in regulating the growth and reproductive function of organisms. There is growing concern that exposure to some chlorinated hydrocarbons such as DTT may affect hormone balance and increase the risk for cancers and reproductive dysfunction. For example, O', P'-DDT, which comprises up to 20% of technical DDT, has been found to compete with the female hormone estradiol for binding to estrogen receptors (3). Environmental chemicals that may perturb hormonal (endocrine) systems have been termed endocrine disrupters.
SUMMARY
Traditional insecticides have targeted nervous system functions that are common to a wide range of species. The use of these insecticides has been associated with the killing of beneficial insects and a risk of toxicity to animals and humans from environmental exposures. Bisacylhydrazine compounds display highly selective insecticidal activity by exploiting subtle differences in the hormonal regulation of insect growth. The novel mechanism of action of these compounds has identified a promising new direction in the development of insecticides that should continue to result in chemicals which have less impact on the environment and are presumably safer to humans. However, the possible relationships between xenobiotics and human disease are very complex. Biochemical changes underlying acute toxicity may be wholly different from more subtle changes that are increasingly recognized as relevant to long-term health. Acute and short-term animal toxicity testing may be grossly inadequate at predicting the long-term impact of chemical exposure on human health. Thus, the continued advancement towards more selective and safer insecticides may be expedited by a combination of 1) targeting insect physiology and 2) screening the effects of candidate compounds on the subcellular processes that are related to human health and disease.
QUESTIONS
1. Distinguish between excitatory and inhibitory neurotransmitters. List the ways by which a xenobiotic might produce acute excitotoxicity. How do lindane and the cyclodiene insecticides produce excitotoxicity?
2. Where is the primary site of insecticidal action of DTT? What factor is believed to account for the greater toxicity of DTT to fish than to mammals?
3. What role does biotransformation play in the acute sensitivities of an organism to organophosphorous insecticides?
4. Are repeated exposures of a crop worker to subtoxic levels of carbamate or organophosphorous insecticides over the course of one to two weeks more likely to result in symptoms of acetylcholine toxicity? Explain.
5. What two characteristics of the bisacylhydrazine insecticides are considered to underlie the ability of these insecticides to induce a lethal molt?
6. Give two reasons why inhibiting mitochondrial electron transport can produce cellular toxicity.
7. How might differences in the levels and activity of cytochrome P450 monooxygenase be involved in the sensitivity to insecticides?
REFERENCES
1. Le Couteur, D.G., McLean, A.J., Taylor, M.C., Woodham, B.L., and Board, P.G. (1999) Pesticides and Parkinson's
disease. Biomed. Pharmacother. 53, 122-130.
2. Taylor, P. and Brown, J.H. (1994) Acetylcholine. In Basic Neurochemistry 5th ed. (Siegel, G.J., Agranoff, B.W.,
Albers, R.W., and Molinoff, P.B., Eds.) pp 231-260, Raven Press, New York.
3. Crosby, D.G. (1998) Environmental Toxicology and Chemistry, Oxford University Press, New York.
4. Timbrell, J. (2000) Principles of Biochemical Toxicology 3rd ed., Taylor & Francis, London.
5. Dhadialla, T.S., Carlson, G.R., and Le, D.P. (1998) New insecticides with ecdysteroidal and juvenile hormone activity.
Annu. Rev. Entomol. 43, 545-569.
6. Wing, K.D. (1988) RH 5849, a nonsteroidal ecdysone agonist: effects on a drosophila cell line. Science 241, 467-469.
7. Wing, K.D., Slawecki, R.A., and Carlson, G.R. (1988) RH 5849, a nonsteroidal ecdysone agonist: effects on larval
Lepidoptera. Science 241, 470-472.
8. Salgado, V.L. (1998) Block of neuronal voltage-dependent K+ channels by diacylhydrazine insecticides.
Neurotoxicology 19, 245-252.
9. Nelson, D.L., and Cox, M.M. (2000) Lehninger Principles of Biochemistry 3rd ed., Worth Publishers, New York.
10. Beal, M.F. (1998) Mitochondrial dysfunction in neurodegenerative diseases. Biochim. Biophys. Acta 1366, 211-223.
11. Carlson, K., and Ehrich, M. (1999) Organophosphorous compound-induced modification of SH-SY5Y human
neuroblastoma mitochondrial transmembrane potential. Toxicol. Appl. Pharmacol. 160, 33-42.
12. Gassner, B., Wuthrich, A., Scholtysik, G., and Solioz, M. (1997) The pyrethroids permethrin and cyhalothrin are potent
inhibitors of the mitochondrial complex I. J. Pharmacol. Exp. Ther. 281, 855-860.
13. Hodgson, E., and Levi, P.E. (1996) Pesticides: an important but underused model for the environmental health sciences.
Environ. Health Perspect.104, 97-106.
14. Butler, A.M., and Murray, M. (1997) Biotransformation of parathion in human liver: participation of CYP3A4 and its
inactivation during microsomal parathion oxidation. J. Pharmacol. Exp. Ther. 280, 966-973.